Иногда в литературе можно встретить диагноз «механобуллезная болезнь». Но в справочниках дерматологии ее нет. На самом деле речь идет о буллезном эпидермолизе – гетерогенной группе генетических патологий, обусловленных мутациями определенных генов, которые ответственны за синтез структурных белков кожи. Их основной признак — появление на коже и слизистых пузырей с серозным содержимым при малейшей механической травме. В последующем на месте буллезной сыпи образуются долго незаживающие эрозии.
Содержание
Классификация
В зависимости от глубины поражения различают три основные формы заболевания и более 30 подтипов патологии, отличающихся по генотипу, фенотипу и характеру наследования.
- Простая форма (ПБЭ). Затронуты только верхние слои эпидермиса. Имеет 12 подтипов, наиболее часто встречающиеся — синдромы Вебера-Коккейна (локализованный), Кёбнера (генерализованный), Доулинга-Меары (герпетиформный), более редкие – ПБЭ Огна, ПБЭ с мышечной дистрофией, ПБЭ с пятнистой пигментацией.
- Пограничная форма (ПгБЭ). Дно пузыря расположено на уровне светлой пластинки – поверхностном слое базальной мембраны. Имеет 2 подтипа. Один из них представлен 6 клиническими формами. Наиболее тяжелый тип, который отличается высокой смертностью – подтип Херлитца.
- Дистрофическая форма (ДБЭ) – страдает верхняя часть сосочкового слоя дермы, которая расположена глубже светлой пластинки. Также имеет два подтипа, которые различаются по механизму наследования – доминантный и рецессивный. Последняя разновидность представлена несколькими клиническими формами, наиболее тяжелая из них – подтип Аллопо-Сименса.
- Синдром Киндлера (смешанный буллезный эпидермолиз). Пузыри могут одновременно затрагивать разные уровни кожи. Самая редкая и мало изученная форма заболевания.
В настоящее время эта классификация считается условной, поскольку на практике встречается очень много клинических форм, из-за чего часто возникают трудности с диагностикой. Специалисты стараются разработать более структурированную и приемлемую классификацию болезни, но пока безуспешно.
Причина
Заболевание развивается вследствие мутации в более чем десяти генах, в которых закодированы белки, составляющие структуру кожи. В большинстве случаев оно передается по наследству, реже является приобретенным, когда изменения в генах происходят спонтанно.
Ребенок может унаследовать патологию от одного из родителей. И даже если они не страдают буллезным эпидермолизом, взрослые могут быть скрытыми носителями мутировавших генов.
Условно все причины разделяют на две группы – генетические и внутриутробные. Основной патогенез – сбой в ферментной системе, в результате чего некоторые протеины становятся мишенью для атаки ферментов.
Генетические нарушения очень разнообразны:
- мутации в генах PLEC, KRT5 и KRT14 вызывают простую форму заболевания, атаке подвергаются протеины базальной мембраны (альфа6-бета4-интегрин) и белки десмосом базального слоя эпидермиса – десмоплакин, плакофиллин-1;
- мутации в генах LAMB3, LAMA3, LAMC2 провоцируют развитие пограничной формы патологии, мишенью становятся коллаген 17-го типа и ламинин-332;
- мутации в гене COL7A1 – причина дистрофической формы, при них происходит повреждение коллагена 7-го типа, который отвечает за стабильность структуры других соединительнотканных волокон кожи;
- мишенью при синдроме Киндлера выступает белок киндлин-1.
Образование пузырей происходит в результате деструкции ключевых структурных белков и нарушения связи между клетками при малейшем механическом воздействии.
Внутриутробные причины, которые могут привести к мутации генов – самые банальные – курение, употребление алкоголя, прием лекарственных препаратов и воздействие других факторов, обладающих тератогенным эффектом.
Формы заболевания и его симптомы
Кожа пациентов не терпит никакого грубого механического или физического воздействия. Иначе в ответ быстро наступает отторжение эпидермиса и дермы. Сначала образуются пузыри, после вскрытия на их месте появляются эрозии. В некоторых случаях к высыпаниям может присоединяться вторичная инфекция.
Врожденный
Врожденный буллезный эпидермолиз может проявляться уже при рождении. В некоторых случаях кожа ребенка травмируется даже в момент прохождения по родовым путям матери.
Однако первые симптомы могут появиться и значительно позже – точных сроков нет. Это может быть период новорожденности, младенчества или раннего детства. Реже всего заболевание начинается в юношестве, но такие случаи также зарегистрированы.
Приобретенный
Приобретенный буллезный эпидермолиз, как уже было сказано выше, возникает в результате спонтанных мутаций. Поэтому чаще всего первые признаки появляются уже у взрослых людей.
Клиническая картина во многом схожа с симптомами этой болезни у детей и зависит исключительно от того, в каких генах произошли изменения.
Простой буллезный эпидермолиз
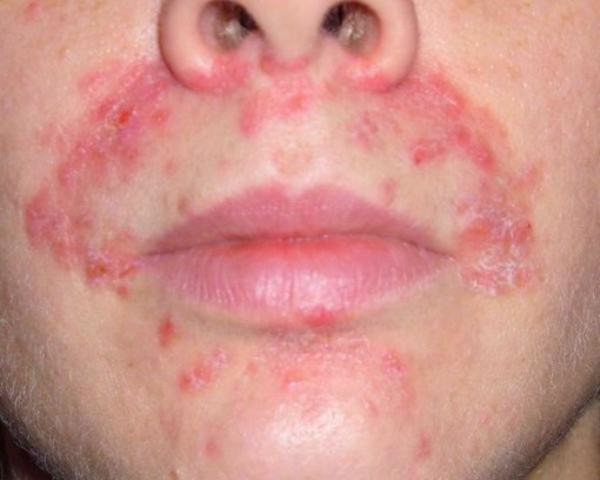
- ПБЭ подтипа Доулинга-Меара (герпетиформный). Наиболее тяжелая форма патологии. Первые признаки видны уже при рождении. Высыпания генерализованные, иногда образуют большие эрозии. Чаще всего поражается слизистая ротовой полости. Патология сопровождается герпетиформными высыпаниями на теле и конечностях. При этом подтипе могут страдать ногти. Происходит их отторжение. Впоследствии отрастают деформированные ногтевые пластинки или длинные ногти с гиперкератозом. Его проявления также возникают на ладонях и подошвах, позже они переходят в кератодермию.


Простой герпетиформный БЭ (Доулинг-Меара) - ПБЭ Кебнера (генерализованный подтип). Обширные высыпания появляются при рождении. Их излюбленная локализация – кисти и стопы. После заживления эрозий остаются участки депигментации. Иногда возникает ладонно-подошвенный гиперкератоз.

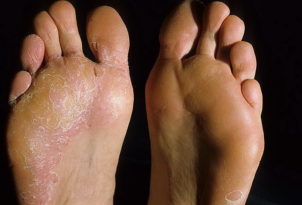
ПБЭ тяжелый генерализованный (Кебнера) - ПБЭ подтипа Вебера-Коккейна (локализованный). Наиболее легкая форма. Впервые заболевание проявляется в младенчестве, реже – в детстве или юношестве. Излюбленная локализация пузырей этого подтипа – ладони и стопы, реже – волосистая часть головы. Незначительные эрозии возникают в ротовой полости. Часто при патологии появляется гипергидроз ладоней и подошв. После заживления эрозий остается пигментация.

ПБЭ локализованный (Вебера-Коккейна) - ПБЭ Огна. Характеризуется сезонным образованием пузырей. Высыпания появляются летом, преимущественно на конечностях. Они сопровождаются онихогрифозом (искривлением ногтя в виде когтя) на больших пальцах ног.
- ПБЭ с мышечной дистрофией. Появление генерализованных пузырей при рождении. Сопровождается прогрессирующей мышечной дистрофией.
- ПБЭ с пятнистой пигментацией. Обширные участки с гиперпигментацией на туловище, кистях рук и стопах. При этом пузырей образуется немного, но по мере их регресса пигментация может усиливаться. Дополнительным симптомом этого подтипа являются веррукозные папулы – бородавчатые наросты на коже. Слизистая ротовой полости страдает незначительно.
Пограничный буллезный эпидермолиз
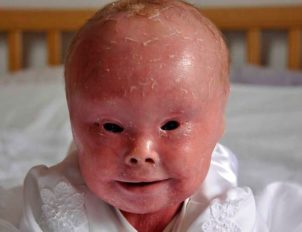
- ПгБЭ Гертлица. Самая тяжелая форма буллезного эпидермолиза. Она приводит к летальному исходу в младенчестве или в раннем детском возрасте. Первые проявления – это обширные пузыри, которые возникают при рождении ребенка. Позже появляются участки гипертрофии грануляционной ткани, преимущественно в области рта, глаз, ноздрей, волосистой части головы и ушных раковин. Дополнительный симптом этого подтипа – утрата ногтевых пластинок и образование гипертрофической грануляционной ткани на концевых фалангах. Страдает ротовая полость – ямки в зубной эмали, обширные эрозии на слизистой ротоглотки. Возможно поражение эпителия носовой полости, слизистой конъюнктивы, пищевода, трахеи, гортани, прямой кишки и уретры. В тяжелых случаях развиваются многочисленные системные нарушения. Заболевание протекает на фоне анемии, происходит задержка роста и физического развития. К летальному исходу приводит осложнение патологии — сепсис.

Пограничный генерализованный тяжелый ВБЭ (летальный Херлитца) - ПгБЭ не-Гертлица. Высыпания в ротовой полости и поражение дыхательных путей не столь тяжелое, как при предыдущем подтипе. Однако встречаются тяжелые аномалии эпителиальной адгезии («склеивания»), что требует проведения трахеотомии с последующей установкой трахеостомы. Наиболее распространенные симптомы – участки сыпи на волосистой части головы и дистрофия ногтевых пластинок, а также периорифициальные (расположенные вокруг естественных отверстий организма, чаще всего – вокруг ануса) эрозии.

ПогрБЭ генерализованный среднетяжелый (не Херлитца) - Локализованный ПгБЭ (минимальный). Течение заболевания довольно легкое. Высыпания локализуются на кистях, стопах и передней поверхности голени. Реже развивается дистрофия ногтей, и появляются небольшие углубления на зубной эмали. На слизистой ротовой полости и носа образуются мелкие эрозии. Прогноз благоприятный.
- ПгБЭ с атрезией (отсутствием или заращением) привратника. Еще одна тяжелая форма заболевания. Пациенты страдают от неимоверной хрупкости кожных покровов и слизистых. У них часто обнаруживают аномалии развития органов мочевыделения, например, гидронефроз или нефрит. Отличительная особенность – рудиментарные ушные раковины.
Дистрофический буллезный эпидермолиз
- Локализованный доминантный ДБЭ (ДДБЭ, подтип Коккейна-Турена). Пузыри образуются на участках, которые чаще всего подвержены трению – колени, область крестца и акральные поверхности. Впоследствии на месте сыпи кожа подвергается дистрофическим процессам, рубцеванию с образованием милиумов. Страдают ногти, вплоть до полного отторжения и атрофического рубцевания последних фаланг пальцев.
- Генерализованный доминантный ДБЭ (ДДБЭ, подтип Пазини). Течение патологии более тяжелое, чем у локализованной формы. Пузыри сменяются эрозиями. После их заживления образуются рубцовые бляшки и милиумы. Дополнительный симптом – спонтанное появление на теле бесцветных плотных папул. По мере взросления пациента генерализованные очаги регрессируют в локализованные с преимущественным поражением кожи конечностей. Заболевание сопровождается дистрофическим изменением ногтей, часто приводящим к утрате ногтевой пластинки, и образованием небольших эрозий на слизистой ротовой полости.
- Рецессивный ДБЭ (РДБЭ). Локализованная форма отличается средней степенью тяжести и по симптомам схожа с ДДБЭ Коккейна-Турена. Подтип Аллопо-Сименса – РДБЭ с тяжелым течением. При рождении возникают обширные генерализованные высыпания. Иногда у ребенка развивается масштабное отслоение кожи (синдром Барта). Чередование обострений и периодов нестойкой ремиссии приводит к распространенному рубцеванию, в результате чего часто образуются сгибательные контрактуры. Заболевание сопровождается проявлением псевдосиндактилии – пальцы ребенка находятся в своеобразной «варежке», что может привести к их сращению. Периорифицивального поражения нет, очаги высыпаний локализуются на голове и шее. Особенно страдает волосистая часть головы, где быстро прогрессирует алопеция. Одно из самых серьезных проявлений – обширное поражение слизистой ротоглотки. После эрозий образуются рубцы, которые ограничивают движения языка и мешают нормально открывать рот. На зубной эмали появляются точечные уплотнения, часто приводящие к кариесу и даже полной потере зубов. Очаги патологии на слизистой верхних дыхательных путей провоцируют стеноз, из-за чего пациенту часто приходится ставить трахеостому. Эрозии в пищеводе вызывают образование стриктур. Сочетание всех этих проявлений приводит к недоеданию и как следствие – задержке роста. У таких детей часто развивается анемия.

Рецессивный дистрофический тяжелый генерализованный БЭ (Аллопо-Сименса)
Что такое синдром Киндлера?
Первые признаки видны уже при рождении. Поскольку высыпания одновременно затрагивают различные ультрастуктурные уровни, клинические проявления могут напоминать как дистрофическую, так и пограничную форму буллезного эпидермолиза. В процессе заживления эрозий на коже происходят атрофические изменения.
По мере взросления буллезные высыпания проходят, но развивается пойкилодермия. Это проявления атрофии, гиперкератоза, сосудистых и пигментных нарушений на коже открытых участков тела, то есть тех, которые подвержены инсоляции. И чаще всего они возникают у пациентов с фоточувствительностью. Помимо этого патология затрагивает ногти и может привести к синдактилии пальцев рук и ног.
Со временем течение заболевания осложняется нарушениями со стороны внутренних органов. Возникают воспалительные процессы на слизистой ротовой полости и стриктуры в пищеводе и уретре.

Диагностика
Анамнез и клиническая картина позволяют установить только предварительный диагноз. Окончательно подтвердить буллезный эпидермолиз могут только методы лабораторной диагностики. Для этого проводят биопсию и образцы кожи подвергают ряду специфических тестов.
- Трансмиссионная электронная микроскопия. Помогает увидеть и оценить определенные элементы кожи, а также обнаружить в ней ультраструктурные изменения.
- Иммунофлюорисцентное антигенное картирование (ИАК). Определяет уровень образования пузыря. Позволяет оценить количество структурных белков и определить, какой из них стал мишенью, а также уточнить ферментную активность ткани. Уменьшение уровня ключевого протеина может свидетельствовать о его низком синтезе или массовом разрушении.
- Генетический анализ (молекулярная или ДНК-диагностика). Завершающий анализ. Прямое секвенирование (определение последовательности соединения нуклеотидов или аминокислот в цепи ДНК или РНК) дает возможность точно определить форму буллезного эпидермолиза и его тип наследования.
Если в семье есть родственники, страдающие буллезным эпидермолизом, целесообразно пройти генетическую консультацию на стадии планирования беременности, а также пренатальную диагностику, которая позволит выявить патологию на раннем этапе развития плода.
Методы лечения
Буллезный эпидермолиз – системная патология, поэтому лечением занимается мультидисциплинарная команда врачей.

Однако современные методы симптоматической терапии, правильный уход за ранами, а также превентивные меры значительно облегчают состояние пациентов и дают им возможность прожить довольно долго.
Цель всех лечебных мероприятий:
- предупреждение разрастания и вскрытия пузырей;
- быстрое заживление эрозий и эпителизация кожи;
- предупреждение развития осложнений.
В качестве симптоматической терапии могут быть назначены следующие группы препаратов:
- анальгетики устранят боль;
- антигистаминные избавят от зуда и жжения;
- антибиотики помогут при вторичном инфицировании эрозий;
- глюкокортикоиды снимут воспаление и неприятные ощущения на коже;
- поливитамины окажут общеукрепляющее действие.

Наружное лечение и уход за кожей заключается в обработке пузырей антисептиками, наложении повязок, в основе которых неприлипающие материалы и несколько слоев бинтов. Кроме того, проводят УФО – профилактическое облучение кожи ультрафиолетовыми лучами для предупреждения инфицирования эрозий.
Продолжительность жизни с буллезным эпидермолизом
Прогноз и продолжительность жизни больного буллезным эпидермолизом зависит от многих факторов — формы патологии и ее подтипа, качества ухода, наличия осложнений и общего состояния пациента.
Простой буллезный эпидермолиз и некоторые локализованные формы других типов имеют наиболее благоприятный прогноз. Иногда проявления болезни регрессируют, и пациенты проживают обычную жизнь. Добиваясь стойкой ремиссии, они никогда не забывают о мерах профилактики и соблюдают все рекомендации врачей.
Наиболее неблагоприятный прогноз при дистрофической форме заболевания. Множественные осложнения часто приводят к летальному исходу еще в детстве.
В настоящее время, к сожалению, не все родители могут обеспечить ребенку-бабочке надлежащий уход. Однако специальные благотворительные фонды не оставляют малышей и помогают им по мере возможности.






Впервые слышу о таком страшном заболевании, которое может повлечь за собой даже смерть. У самое ужасное что от него не лекарства. Прочитав статью я понял что оно очень редкое, но все же встречается а особенно у новорожденных.